Note
This page was generated from scgen_perturbation_prediction.ipynb. Interactive online version: ![]() .
.
SCGEN: Perturbation Prediction¶
[1]:
import sys
#if branch is stable, will install via pypi, else will install from source
branch = "stable"
IN_COLAB = "google.colab" in sys.modules
if IN_COLAB and branch == "stable":
!pip install --quiet scgen[tutorials]
elif IN_COLAB and branch != "stable":
!pip install --quiet --upgrade jsonschema
!pip install --quiet git+https://github.com/theislab/scgen@$branch#egg=scgen[tutorials]
[2]:
import logging
import scanpy as sc
import scgen
Loading Train Data¶
[3]:
train = sc.read("./tests/data/train_kang.h5ad",
backup_url='https://drive.google.com/uc?id=1r87vhoLLq6PXAYdmyyd89zG90eJOFYLk')
/home/marco/.pyenv/versions/3.7.7/lib/python3.7/site-packages/anndata/compat/__init__.py:183: FutureWarning: Moving element from .uns['neighbors']['distances'] to .obsp['distances'].
This is where adjacency matrices should go now.
FutureWarning,
/home/marco/.pyenv/versions/3.7.7/lib/python3.7/site-packages/anndata/compat/__init__.py:183: FutureWarning: Moving element from .uns['neighbors']['connectivities'] to .obsp['connectivities'].
This is where adjacency matrices should go now.
FutureWarning,
Let’s remove stimulated CD4T cells from both the training set. This is just for the sake of this notebook, in practice, you do not need to do this step, just pass the train data
[4]:
train_new = train[~((train.obs["cell_type"] == "CD4T") &
(train.obs["condition"] == "stimulated"))]
Preprocessing Data¶
[6]:
scgen.SCGEN.setup_anndata(train_new, batch_key="condition", labels_key="cell_type")
INFO Using batches from adata.obs["condition"]
INFO Using labels from adata.obs["cell_type"]
INFO Using data from adata.X
INFO Computing library size prior per batch
INFO Successfully registered anndata object containing 13766 cells, 6998 vars, 2 batches,
7 labels, and 0 proteins. Also registered 0 extra categorical covariates and 0 extra
continuous covariates.
INFO Please do not further modify adata until model is trained.
/home/marco/.pyenv/versions/scgen_test/lib/python3.7/site-packages/scvi/data/_anndata.py:815: UserWarning: adata.X does not contain unnormalized count data. Are you sure this is what you want?
logger_data_loc
/home/marco/.pyenv/versions/scgen_test/lib/python3.7/site-packages/pandas/core/arrays/categorical.py:2487: FutureWarning: The `inplace` parameter in pandas.Categorical.remove_unused_categories is deprecated and will be removed in a future version.
res = method(*args, **kwargs)
/home/marco/.pyenv/versions/scgen_test/lib/python3.7/site-packages/pandas/core/arrays/categorical.py:2487: FutureWarning: The `inplace` parameter in pandas.Categorical.remove_unused_categories is deprecated and will be removed in a future version.
res = method(*args, **kwargs)
Creating and Saving the model¶¶
[7]:
model = scgen.SCGEN(train_new)
model.save("../saved_models/model_perturbation_prediction.pt", overwrite=True)
WARNING Make sure the registered X field in anndata contains unnormalized count data.
Training the Model¶
[8]:
model.train(
max_epochs=100,
batch_size=32,
early_stopping=True,
early_stopping_patience=25
)
GPU available: True, used: True
TPU available: None, using: 0 TPU cores
LOCAL_RANK: 0 - CUDA_VISIBLE_DEVICES: [0]
Epoch 31/100: 31%|███ | 31/100 [01:54<04:14, 3.68s/it, loss=74.7, v_num=1]
Latent Space¶
[9]:
latent_X = model.get_latent_representation()
latent_adata = sc.AnnData(X=latent_X, obs=train_new.obs.copy())
WARNING Make sure the registered X field in anndata contains unnormalized count data.
[10]:
sc.pp.neighbors(latent_adata)
sc.tl.umap(latent_adata)
sc.pl.umap(latent_adata, color=['condition', 'cell_type'], wspace=0.4, frameon=False,
save='latentspace_batch32_klw000005_z100__100e.pdf')
WARNING: You’re trying to run this on 100 dimensions of `.X`, if you really want this, set `use_rep='X'`.
Falling back to preprocessing with `sc.pp.pca` and default params.
WARNING: saving figure to file figures/umaplatentspace_batch32_klw000005_z100__100e.pdf
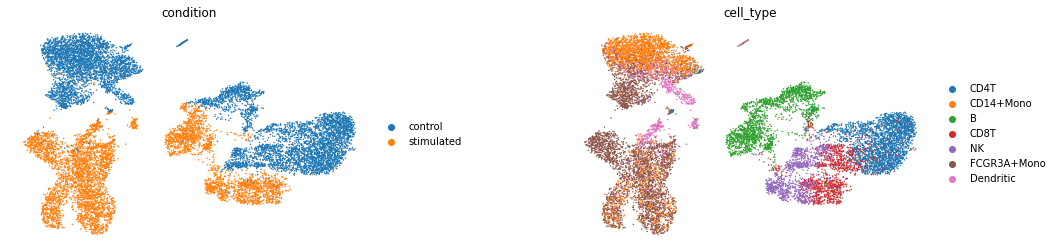
Prediction¶
After training the model you can pass the adata of the cells you want to perturb. Here we pass unperturbed CD4T cells
Here the ‘adata’ contains the cells that you want estimate the perturbation based on them. we set “ctrl” to our control labels and “stim” to our stimulated labels. If you apply it in another context just set “ctrl” :”your_control_label” and “stim”:”your_stimulated_label”. the returned value is a numpy matrix of our predicted cells and the second one is the difference vector between our conditions which might become useful later.
[11]:
pred, delta = model.predict(
ctrl_key='control',
stim_key='stimulated',
celltype_to_predict='CD4T'
)
pred.obs['condition'] = 'pred'
/home/marco/.pyenv/versions/scgen_test/lib/python3.7/site-packages/pandas/core/arrays/categorical.py:2487: FutureWarning: The `inplace` parameter in pandas.Categorical.remove_unused_categories is deprecated and will be removed in a future version.
res = method(*args, **kwargs)
Observation names are not unique. To make them unique, call `.obs_names_make_unique`.
Observation names are not unique. To make them unique, call `.obs_names_make_unique`.
INFO Received view of anndata, making copy.
Observation names are not unique. To make them unique, call `.obs_names_make_unique`.
WARNING Make sure the registered X field in anndata contains unnormalized count data.
INFO Received view of anndata, making copy.
Observation names are not unique. To make them unique, call `.obs_names_make_unique`.
WARNING Make sure the registered X field in anndata contains unnormalized count data.
INFO Received view of anndata, making copy.
WARNING Make sure the registered X field in anndata contains unnormalized count data.
In the previous block, the difference between conditions is by default computed using all cells (obs_key=”all”). However, some times you might have a rough idea that which groups (e.g. cell types) are close to your cell type of interest. This might give you more accurate predictions. For example, we can restrict the delta computation only to CD8T and NK cells. We provide dictionary in form of obs_key={“cell_type”: [“CD8T”, “NK”]} which is telling the model to look at “cell_type” labels in adata (here: train_new) and only compute the delta vector based on “CD8T” and “NK” cells :
pred, delta = scg.predict(adata=train_new, adata_to_predict=unperturbed_cd4t, conditions={“ctrl”: “control”, “stim”: “stimulated”}, cell_type_key=”cell_type”, condition_key=”condition”, obs_key={“cell_type”: [“CD8T”, “NK”]})`
Evaluation of the predcition¶¶
Extracting both control and real stimulated CD4T cells from our dataset¶
[12]:
ctrl_adata = train[((train.obs['cell_type'] == 'CD4T') & (train.obs['condition'] == 'control'))]
stim_adata = train[((train.obs['cell_type'] == 'CD4T') & (train.obs['condition'] == 'stimulated'))]
Merging predicted cells with real ones
[13]:
eval_adata = ctrl_adata.concatenate(stim_adata, pred)
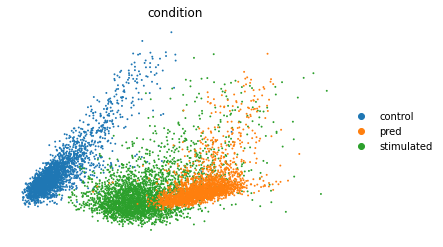
Embedding all real and predicted cells in one PCA plot¶¶
[14]:
sc.tl.pca(eval_adata)
sc.pl.pca(eval_adata, color="condition", frameon=False,
save='pred_stim_b32_klw000005_z100__100e.pdf')
... storing 'condition' as categorical
WARNING: saving figure to file figures/pcapred_stim_b32_klw000005_z100__100e.pdf
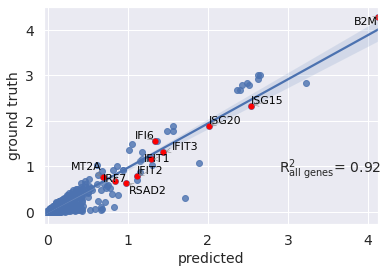
Mean correlation plot¶¶
You can also visualize your mean gene expression of your predicted cells vs control cells while highlighting your genes of interest (here top 10 differentially expressed genes)
[15]:
CD4T = train[train.obs["cell_type"] =="CD4T"]
/home/marco/.pyenv/versions/scgen_test/lib/python3.7/site-packages/pandas/core/arrays/categorical.py:2487: FutureWarning: The `inplace` parameter in pandas.Categorical.remove_unused_categories is deprecated and will be removed in a future version.
res = method(*args, **kwargs)
[16]:
sc.tl.rank_genes_groups(CD4T, groupby="condition", method="wilcoxon")
diff_genes = CD4T.uns["rank_genes_groups"]["names"]["stimulated"]
print(diff_genes)
Trying to set attribute `.uns` of view, copying.
['ISG15' 'IFI6' 'ISG20' ... 'FTL' 'RGCC' 'FTH1']
[17]:
r2_value = model.reg_mean_plot(
eval_adata,
axis_keys={"x": "pred", "y": "stimulated"},
gene_list=diff_genes[:10],
labels={"x": "predicted", "y": "ground truth"},
path_to_save="./reg_mean1.pdf",
show=True,
legend=False
)
/home/marco/.pyenv/versions/scgen_test/lib/python3.7/site-packages/pandas/core/arrays/categorical.py:2487: FutureWarning: The `inplace` parameter in pandas.Categorical.remove_unused_categories is deprecated and will be removed in a future version.
res = method(*args, **kwargs)
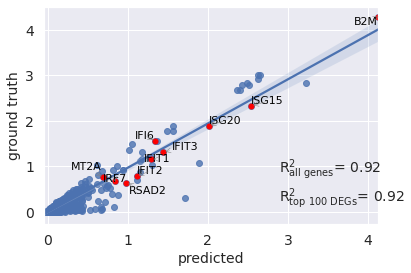
You can also pass a list of differentially epxressed genes to compute correlation based on them
[18]:
r2_value = model.reg_mean_plot(
eval_adata,
axis_keys={"x": "pred", "y": "stimulated"},
gene_list=diff_genes[:10],
top_100_genes= diff_genes,
labels={"x": "predicted","y": "ground truth"},
path_to_save="./reg_mean1.pdf",
show=True,
legend=False
)
/home/marco/.pyenv/versions/scgen_test/lib/python3.7/site-packages/pandas/core/arrays/categorical.py:2487: FutureWarning: The `inplace` parameter in pandas.Categorical.remove_unused_categories is deprecated and will be removed in a future version.
res = method(*args, **kwargs)
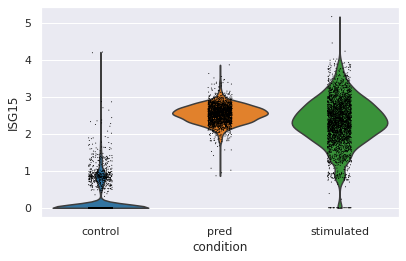
Violin plot for a specific gene¶¶
Let’s go deeper and compare the distribution of “ISG15”, the top DEG between stimulated and control CD4T cells between predcited and real cells
[19]:
sc.pl.violin(eval_adata, keys="ISG15", groupby="condition")
[ ]: